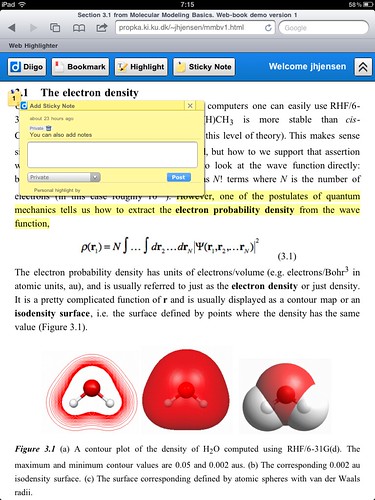
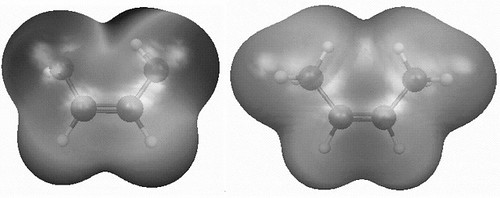
Recently a colleague of mine, Henrik, lamented that at a recent oral exam many first year students had no clue what a typical bond length in a molecule is. I am not a big fan of memorizing something that can be easily Googled, but we're talking order of magnitude here, not the last decimal place.
This is a place where molecular modeling tools such as Avogadro can help, but how to do this exactly? Knowing an approximate bond length by heart comes from looking at many, many molecular structures, and imagining a worksheet with dozens of fill-in-the-blank bond lengths already puts me to sleep.
But how about something like: use Avogadro to build a molecule with the longest CC single bond possible. This sounds a bit more fun and I bet a few strategies are already popping into your mind.
Based on my recent excellent experience with Google Docs, I would then have student collect answers on a common Google spreadsheet. Students could either collect their work on a single sheet or one sheet per student/team. This allows students to learn different design principles from each other and adds an element of competition (gamification) since you can now compare results.
Would this actually work? I challenge you to find out!
By work I mean: engage students enough to build a large number of molecules and think about bond lengths? I don't know, but how about testing it out here? I have started a spreadsheet with a few entries. If you can do better, feel free to add your entries.
Some ground rules:
1. The bond length must come from the lowest energy conformation
2. To beat a current entry the CC bond length must be longer by 0.005 Å
3. You must use the MMFFs force field (which will limit the number of elements at your disposal).
4. For now I'll leave the maximum number of atoms open, but the fewer atoms you use, the more impressed I will be.
You might have the students figure these issues out by themselves. Explaining why they are important could be very teachable moments.
Practical tips
1. As the molecules get more complicated, naming them becomes difficult. In that case I suggest using SMILES, which you can generate with this GUI. It would be nice if Avogadro has a "Copy SMILES" option.
2. You can build your molecule using SMILES.
This is a place where molecular modeling tools such as Avogadro can help, but how to do this exactly? Knowing an approximate bond length by heart comes from looking at many, many molecular structures, and imagining a worksheet with dozens of fill-in-the-blank bond lengths already puts me to sleep.
But how about something like: use Avogadro to build a molecule with the longest CC single bond possible. This sounds a bit more fun and I bet a few strategies are already popping into your mind.
Based on my recent excellent experience with Google Docs, I would then have student collect answers on a common Google spreadsheet. Students could either collect their work on a single sheet or one sheet per student/team. This allows students to learn different design principles from each other and adds an element of competition (gamification) since you can now compare results.
Would this actually work? I challenge you to find out!
By work I mean: engage students enough to build a large number of molecules and think about bond lengths? I don't know, but how about testing it out here? I have started a spreadsheet with a few entries. If you can do better, feel free to add your entries.
Some ground rules:
1. The bond length must come from the lowest energy conformation
2. To beat a current entry the CC bond length must be longer by 0.005 Å
3. You must use the MMFFs force field (which will limit the number of elements at your disposal).
4. For now I'll leave the maximum number of atoms open, but the fewer atoms you use, the more impressed I will be.
You might have the students figure these issues out by themselves. Explaining why they are important could be very teachable moments.
Practical tips
1. As the molecules get more complicated, naming them becomes difficult. In that case I suggest using SMILES, which you can generate with this GUI. It would be nice if Avogadro has a "Copy SMILES" option.
2. You can build your molecule using SMILES.